The Medical Industry’s Move Toward Quality, Part
Two
 In
my February column, I discussed the Food and Drug Administration’s
plans to introduce a risk-based approach to its projected
21st century update of the current Pharmaceutical Good Manufacturing
Practice. To better understand what this approach might
entail, one must look at what’s currently taking place
in the medical device industry, which is already moving
toward a risk-based approach. In
my February column, I discussed the Food and Drug Administration’s
plans to introduce a risk-based approach to its projected
21st century update of the current Pharmaceutical Good Manufacturing
Practice. To better understand what this approach might
entail, one must look at what’s currently taking place
in the medical device industry, which is already moving
toward a risk-based approach.
Risk management is hardly new. It’s academically
defined as “the techniques used to minimize and prevent
accidental loss to a business.” This definition first
appeared between 1960 and 1965.
The aerospace and defense industries have been using risk
analysis and evaluation techniques for decades as an integral
part of the design process. Methodologies such as failure
mode and effects analysis, failure mode effect and criticality
analysis, fault tree analysis and reliability-centered maintenance
are well-established design and process review tools in
these industries.
Moreover, risk-based initiatives are not new to the FDA.
Categorizing medical devices into Classes I, II and III--based
on ascending levels of potential harm to the patient and
with increasingly stringent standards for each class--is
a risk-based approach. Hazard analysis and critical control
points applied to the food proc-essing industry represent
an example of the FDA imposing a risk-assessment requirement
on a specific industry sector.
New in the medical device industry is the recent publication
of ISO 13485:2003, Medical Devices--Quality management systems,
requirements for regulatory purposes. This standard incorporates
several significant and interrelated changes to the 1996
version as well as to ISO 9001:2000, the standard upon which
ISO 13485:2003 is based. The new version includes:
 The addition of the phrase “for regulatory purposes”
The addition of the phrase “for regulatory purposes”
A stated objective of promoting “harmonized medical
device regulatory requirements”
A requirement for a “process approach to quality management”
The inclusion of many documentation and record requirements
dropped by ISO 9001:2000
The addition of documented requirements for risk management
Canada and the European Union recognize ISO 13485:2003,
whereas the FDA, for now, is sticking with its Quality System
Regulation. Therefore, manufacturers in the United States
will need quality management systems that cover both (not
two separate management systems, as some have suggested).
The deadline to transition from ISO 13485:1996 to the 2003
version is July 15, 2006. However, organizations doing business
in Canada must transition by March 15, 2006. The most significant
difference between the two versions is the risk management
requirement.
To begin, the standard requires documented risk management
throughout the product realization cycle, which includes
product planning, determining customer requirements, design
and development, production and service, and control of
monitoring and measuring devices. Some key processes included
are design verification and validation, verification of
purchased product, and validation of production and service
products. If there’s any doubt as to ISO 13485:2003’s
intent, the standard refers users to ISO 14971:2000, Medical
devices--Application of risk management to medical devices.
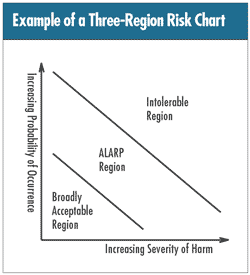
According to ISO 14971:2000, “risk” in medical
devices lingo is a “combination of the probability
of the occurrence of harm and the severity of that harm.”
The higher the probability of occurrence and/or the greater
the severity of potential harm, the lower the acceptability
of risk. The accompanying figure, adopted from ISO 14971:2000,
Annex E, illustrates the concept. (Note: the acronym ALARP
means “as low as reasonably practicable.”)
Risk management--or the “systematic application
of management policies, procedures and practices to the
tasks of analyzing, evaluating and controlling risk”--comprises
four stages:
1. Risk analysis
2. Risk evaluation
3. Risk control
4. Postproduction information
The first two--analysis and evaluation--compose risk assessment,
which is an essential component of product planning. ISO
14971:2000 further breaks down the four stages into 13 detailed
steps, each of which must be documented and recorded to
verify compliance.
Clearly, a risk-based approach is the logical extension
of a process approach to quality management systems. In
auditing management systems, both internally and externally,
organizations must concentrate on processes most essential
to the final product’s quality and safety. If the
final product is a medical or pharmaceutical device, its
potential to cause harm is a significant characteristic
that must be assessed and monitored.
In practical terms, a risk- or process-based approach
means that medical device and pharmaceutical developers,
manufacturers, certification bodies and regulatory agencies
must have auditors and inspectors who can:
Understand process auditing and are capable of reviewing
and understanding risk analysis methodologies
Evaluate the adequacy of an organization’s risk management
files
Process validation reports and other critical data
Verify that an organization is in compliance with ISO standards
and federal regulations
We must truly move from cursory audits of all processes
to in-depth audits of critical processes. It’s a big
job.
Stanley A. Marash, Ph.D., is chairman and CEO of The
SAM Group, which includes STAT-A-MATRIX Inc. and Oriel Inc.
Marash is the author of Fusion Management (QSU Publishing
Co., 2003). Note: Fusion Management is a trademark of STAT-A-MATRIX
Inc. ©2004 STAT-A-MATRIX Inc. All rights reserved.
|
